GAAP is a cGOF (core-gene-defined Genome-organization-framework) Assisted Assembly Pipeline. It is aimed at scaffolding and extending scaffolds and contigs based on de novo assembly of one paired-end library and core gene cluster from multiple related references.
GAAP is composed of two separate yet sequential sections:
1) cGOF_identification, which extracts sequences and order & orientation of cGOF segments from references; one species run once.
2) Scaffolding, which uses segments of cGOF genes as anchors to order the target scaffolds and contigs, uses paired-end reads mapping for local scaffolding of ordered scaffolds/contgis to recover more contigs, and then matches the closest organized reference to construct a pseudogenome; one target run once.
The framework and algorithm of GAAP are shown in Fig.1.
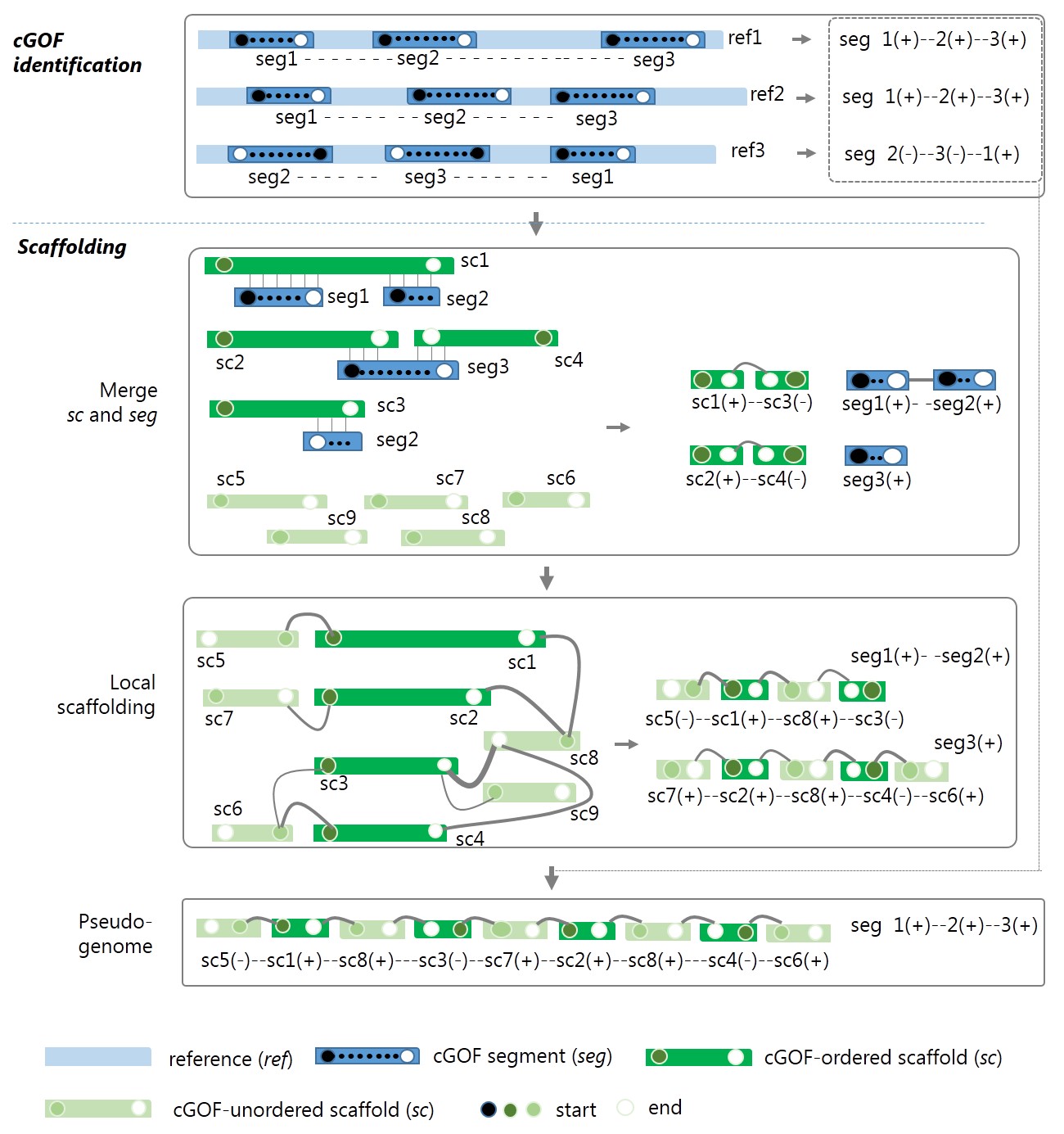
Figure 1 The framework of GAAP. seg, segment of cGOF; ref, reference; sc, scaffold/contig. Head (closed circle) and tail (open circle) vertices of the syntenic seg in each reference are sequentially connected with a dashed line indicating the permutation (order and orientation) of seg. The graph in the local scaffolding of ordered sc is built by connecting seg-ordered sc and unordered sc, where the links are higher than a certain cut-off. If a conflicted connection occurs, the priorities are seg-ordered by the order and link count. The line widths indicate the link count.The line widths indicate the link count. For each pair of scaffolds and contigs, sci and scj, there exist four types of connection between them, (i) head-to-head, [sci(-),scj(+)] or [scj(-),sci(+)]; (ii)head-to-tail, [sci(-),scj(-)] or [scj(-),sci(-)]; (iii) tail-to-head, [sci(+),scj(+)] or [scj(+),sci(+)]; (iv) tail-to-tail, [sci(+),scj(-)] or [scj(+),sci(-)]; where positive and negative signs indicate the relative orientation of assemblies.
GAAP is in Python scripts (Python version of 2.7 or above is required to run the program), so it is unnecessary to compile. However, extra programs Bowtie2 and BLAT are required to run GAAP. Bowtie2 is available from http://bowtie-bio.sourceforge.net/bowtie2/index.shtml. BLAT is available from ftp://ftp.ncbi.nih.gov/blast/executables/release/.
Before start, put Bowtie2 and BLAT to your $PATH: 'export PATH=path-to-bowtie2:$PATH' and 'export PATH=path-to-blat:$PATH'.
Besides, PGAP (http://sourceforge.net/projects/pgap/) is recommended to produce gene cluster file, which is needed to run cGOF identification.
1) Paired-end reads in FASTA or FASTQ format.
2) Contigs or scaffolds assembled by de novo DNA-Seq assembler (SOAPdenovo, Velvet, ABySS, ALLPATHS-LG, etc.).
3) Gene cluster files generated from genomes of a closely related species (no less than ten genomes are recommended), including ortholog cluster file and corresponding ptt files and nucleotide sequences from NCBI ftp. See "examples" for detail.
1) cGOF identification
python cgof_identification.py [-h] [-s SEGMENT_LENGTH] [-o OUTPUT_DIR] NAME CLUSTER_FILE PTT_DIR REFERENCE_FILE
Inputs:
Outputs:
Options:
2) Scaffolding
python scaffolding.py [-h] [-r R] [-m MAX_INSERT] [-n MIN_INSERT] [-s SCAFFOLD_SIZE] [-o BLAT_OUTPUT] [-c] READS_1 READS_2 SCAFFILDS_FASTA GOF_FASTA SEGMENT_FREQUENCY OUTPUT_DIR
Inputs:
Outputs:
Options:
Here we provide two examples, S.aureus and S.suis.
1) For S.aureus, we run command lines as follows.
In this step, GAAP takes files as follows as input, and outputs sau.output.fasta and sau.output.freq, and the minimum segment length is set to 10 cGOF genes.
CLUSTER_FILE (sau_gene.cluster)
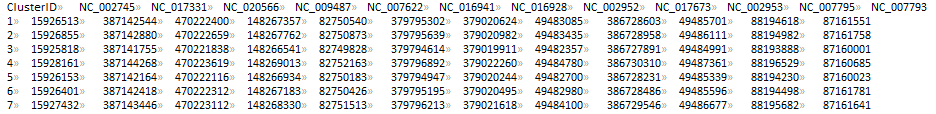
PTT_DIR (ptt_dir/)

REFERENCE_FILE (NC_002745.nuc)

NAME.output.fasta (sau.output.fasta)."S"indicates segment.

NAME.output.freq (sau.output.freq)
python scaffolding.py -m 550 -n 450 -s 300 -o sau -c sau_1.fa sau_2.fa sau_scafseq.fa output/sau.output.fasta output/sau.output.freq output/
In this step, GAAP takes paired-end reads (sau_1.fa and sau_2.fa, and the insert length is 450~550bp), target scaffolds (sau.scafseq, and the minimum length is set to 300bp), sau.output.fasta and sau.output.freq as input, and outputs sau.genome.fasta, sau.segmented.fasta and sau.unsed.fasta in output/ directory.
2) For S.ssuis, we run command lines as follows.
python cgof_identification.py -o output/ ssuis ssuis_gene.cluster ptt_dir/ NC_009442.nuc
In this step, GAAP takes CLUSTER_FILE (ssuis_gene.cluster), PTT_DIR (ptt_dir/) and REFERENCE_FILE (NC_009442.nuc) as input, and outputs ssuis.output.fasta and ssuis.output.freq.
NAME.output.freq (ssuis.output.freq)."S"indicates segment,and "rc" indicates reverse complement.
python scaffolding.py -r 50 -m 550 -n 450 -s 300 -o ssuis -c ssuis_1.fa ssuis_2.fa ssuis_scafseq.fa output/ssuis.output.fasta output/ssuis.output.freq output/
In this step, GAAP takes paired-end reads (ssuis_1.fa and ssuis_2.fa), target scaffolds (ssuis.scafseq), ssuis.output.fasta and ssuis.output.freq as input, and outputs ssuis.genome.fasta, ssuis.segmented.fasta and ssuis.unused.fasta in output/ directory. The insert length of reads is 450~550bp (-m 550 -n 450), the minimum length of scaffolds is set to 300bp (-s 300), and one read pairs are remained in the input reads data if the first 50bp are identical in more than one pairs (-r 50).